![]()
Transcriptomic profiling of skeletal muscle from the Ts1Cje mouse model of Down syndrome suggests dysregulation of trisomic genes associated with neuromuscular junction signaling, oxidative stress and chronic inflammation
Melody Pui Yee Leong 1,2, Usman Bala 2,3, Chai Ling Lim 2,3, Rozita Rosli 1,2, Pike-See Cheah 2,3 and King-Hwa Ling 1,2,*
1 Department of Biomedical Science, Faculty of Medicine and Health Sciences, Universiti Putra Malaysia, 43400 UPM Serdang, Selangor, Malaysia.
2 Genetics and Regenerative Medicine Research Centre (GRMRC), Faculty of Medicine and Health Sciences, Universiti Putra Malaysia, 43400 UPM Serdang, Selangor, Malaysia.
3 Department of Human Anatomy, Faculty of Medicine and Health Sciences, Universiti Putra Malaysia, 43400 UPM Serdang, Selangor, Malaysia.
* Correspondence: lkh@upm.edu.my; Tel.: +603-8947 2564
Received: 10 May 2018; Accepted: 21 August 2018; Published: 27 August 2018
Edited by: Azlina Ahmad Annuar (University of Malaya, Malaysia)
Reviewed by: Ruth Chia (National Institute on Aging, National Institutes of Health, USA); Shelisa Tey (Institute of Neuropathology, RWTH University Hospital Aachen, Germany)
DOI: https://doi.org/10.31117/neuroscirn.v1i1.12
Abstract: Ts1Cje is a mouse model of Down syndrome (DS) with partial triplication of chromosome 16, which encompasses a high number of human chromosome 21 (HSA21) orthologous genes. The mouse model exhibits muscle weakness resembling hypotonia in DS individuals. The effect of extra gene dosages on muscle weakness or hypotonia in Ts1Cje and DS individuals remains unknown. To identify molecular dysregulation of the skeletal muscle, we compared the transcriptomic signatures of soleus and extensor digitorum longus (EDL) muscles between the adult Ts1Cje and disomic littermates. A total of 166 and 262 differentially expressed protein-coding genes (DEGs) were identified in the soleus and EDL muscles, respectively. The partial trisomy of MMU16 in Ts1Cje mice has a greater effect on gene expression in EDL. Top-down clustering analysis of all DEGs for represented functional ontologies revealed 5 functional clusters in soleus associated with signal transduction, development of reproductive system, nucleic acid biosynthesis, protein modification and metabolism as well as regulation of gene expression. On the other hand, only 3 functional clusters were observed for EDL namely neuron and cell development, protein modification and metabolic processes as well as ion transport. A total of 11 selected DEGs were validated using qPCR (disomic DEGs: Mansc1; trisomic DEGs: Itsn1, Rcan1, Synj1, Donson, Dyrk1a, Ifnar1, Ifnar2, Runx1, Sod1 and Tmem50b). The validated DEGs were implicated in neuromuscular junction signalling (Itsn1, Syn1), oxidative stress (Sod1, Runx1) and chronic inflammation processes (Runx1, Rcan1, Ifnar1, Ifnar2). Other validated DEGs have not been well-documented as involved in the skeletal muscle development or function, thus serve as interesting novel candidates for future investigations. To our knowledge, the study was the first attempt to determine the transcriptomic profiles of both soleus and EDL muscles in Ts1Cje mice. It provides new insights on the possible disrupted molecular pathways associated with hypotonia in DS individuals.
Keywords: soleus; extensor digitorum longus; skeletal muscle; microarray; Down syndrome;
©2018 by Leong et al for use and distribution in accord with the Creative Commons Attribution (CC BY-NC 4.0) license (https://creativecommons.org/licenses/by-nc/4.0/), which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original author and source are credited.
1. INTRODUCTION
Down syndrome (DS) is a genetic disorder caused by trisomy of human chromosome (HSA) 21 and is the most common genetic form of intellectual disability. In addition, the disorder is characterised by a collection of more than 80 clinical features affecting almost all organs within the body [1] including characteristic set of facial features, early onset dementia, weak immune system and hypotonia [2]. DS individuals with hypotonia have been shown to have difficulty in postural control [3], instability in trunk control [4], delayed motor function development [5,6], perceptual motor deficits [7] and difficulty in motor planning [8] thus negatively impacting on their occupational and social development as well as quality of life [9-11].
DS individuals demonstrate lower levels of muscle strength as compared to those without DS or intellectual disability [12-14]. In general, although children with DS learn to walk, reach and grasp objects, feed themselves and perform many other fundamental skills, their movements lack in precision, are poorly coordinated and less efficient than the movements of normally developing children [15]. Compared to normal individuals, children and adults with DS are slower and more variable in reaction time tasks [16]. Besides exhibiting clumsy sequences of movements of different parts of the arm and hand, DS individuals also tend to have poorer control in programming actions timing and force reduction [16,17]. Motor development of DS children occurs later than normal children. DS children usually do not walk before the age of two. To compensate for these characteristics, children with DS adopt motor patterns that compensate for what they lack in terms of strength, joint stability and length of their limbs [18]. In the long run, if these patterns are repeated frequently they can lead to abnormalities in the musculoskeletal system and motor disabilities.
The underlying pathology of muscle weakness in DS individuals is not fully understood. Motor development deficits in DS are suggested to be determined by structural and functional alterations of the central nervous system (CNS). Brains of DS individuals have been characterised with hypocellularity of the hippocampus, decreased weight and volume; reduced neuron density especially in the cerebral cortex, hippocampus, cerebellum and brainstem; lower synaptic density and reduced number of neurotransmitters; and delayed myelination [19,20]. These abnormalities in the various brain structures including the midbrain and cerebellum have been linked to muscle tone determination, balance and coordination [21]. Interestingly, reduced cerebellar volume has become a consistent finding in DS individuals [22]. However, despite the differences in the volume of the cerebellum in DS individuals, the reduction is non-progressive and do not further reduce significantly with age [23]. Therefore, age-related decline in fine motor activities in DS is not necessarily the result of reduced cerebellar volume suggesting an impairment of the peripheral innervation or the skeletal muscle of DS individuals also contribute to hypotonia.
The current study reported the transcriptomic analysis on two types of skeletal muscles namely soleus (Type I slow twitch) and extensor digitorum longus (EDL, Type II fast twitch) harvested from a mouse model known as Ts1Cje. We attempted to understand the effect of extra gene dosage on skeletal muscle function in DS due to the triplication of HSA21 genes. Many DS mouse models have been generated exhibiting similar phenotypes seen in DS individuals [24] including motor dysfunctions, such as delayed motor skill development, poor fine motor movement and impaired motor coordination [25-27]. Orthologs of HSA21 genes are dispersed among 3 mouse (MMU) chromosomes namely the telomeric region of MMU16, internal segments of MMU17 and MMU10 [28]. The Ts1Cje mouse model has a triplicated segment of the MMU16 with approximately 85 genes synteny to genes located on HSA21 [2,29]. The model has been shown to exhibit features similar to DS humans such as hypocellularity of the brain, decrease in cognitive ability as well as behavioural and motor abnormalities [30-34]. A recent study showed that besides being genetically representative to DS in humans, the mouse soleus muscle has a great molecular similarity to human skeletal muscles, independent of the anatomic location or muscle type [35] making it a suitable model for studies on molecular characterisation of muscle weakness in Ts1Cje.
Soleus muscle can be found at the calf at beneath the gastrocnemius, originating from the upper two-thirds posterior surfaces of the tibia and fibula. It is a Type I slow twitch muscle that contract slowly but can contract repeatedly for a long period of time resulting in high resistance to fatigue [36]. Thus, it exhibits high mitochondrial numbers and oxidative enzyme content as well as low glycogen levels and glycolytic enzyme activity. On the other hand, Type II fast twitch muscle of Ts1Cje namely extensor digitorum longus (EDL) which is situated at the lateral part of the leg adjacent to the tibialis anterior muscle. Fast twitch muscles have a faster contraction speed but achieves fatigue earlier as compared to slow twitch muscles [36]. Thus, transcriptomic profiling and comparison of these muscles in Ts1Cje mouse model will lead to the identification of dysregulated genes and molecular networks related to muscle weakness due to the partial trisomy of MMU16. The fundamental knowledge of genotype to phenotype (muscle weakness) correlations in Ts1Cje will enhance the current understanding of mechanisms underlying muscle weakness or hypotonia in DS.
2. MATERIALS AND METHODS
2.1 Animal Husbandry & Genotyping
Two groups of adult mice, the Ts1Cje and wild type (disomic C57BL/6) at postnatal day 56-70 were used in the study. Ts1Cje and wild type mice were generated by mating the Ts1Cje males with a wild type C57BL/6 female as described previously [29]. All experimental protocols were performed with the approval from the Institutional Animal Care and Use Committee of Universiti Putra Malaysia (UPM/IACUC/AUP-R003/2014). The disomic littermates served as the control for the study. The mice were identified by tail genotyping using polymerase chain reaction (PCR) to amplify the Grik1 gene in WT mice and both Grik1 and Neo genes in the Ts1Cje mouse as described by [29]. All mice were housed under controlled temperatures with a 12:12 hour light:dark cycle. The mice were given unlimited access to standard animal feed (Altromin 1324, Germany) and sterile water ad libitum.
2.2 Skeletal Muscle Harvesting
The skeletal muscle samples collected in this study were namely soleus and extensor digitorum longus (EDL). The mouse was euthanised by inhalation of 4% mg/kg isoflurane fume chamber followed by cervical dislocation and pinned facing upwards. The muscles were isolated using the tendon to tendon method. The entire length of the hindlimb was exposed by removal of the skin and without damaging the underlying muscles. The EDL was visually located at the anterior compartment of the hindlimb just underneath the tibialis anterior (TA) muscle. The distal tendons of the TA and EDL were cut and both muscles were held by their tendons and pulled gently up towards the proximal end. The EDL muscle was located just underneath the TA muscle by pulling the tendons of the respective muscles in opposite directions. The TA was removed by cutting at the proximal end to expose the EDL proximal tendon. The proximal tendon of the EDL was cut to obtain the muscle. The mouse was then flipped around and pinned face down on the dissection board. The soleus muscle which was visually located at the posterior compartment of the hind limb just underneath the gastrocnemius (GA) muscle was then isolated in similar manner. Caution was taken to ensure that the exposed tissues do not dry out by flushing with ice cold 1X PBS when required. All muscle tissue samples were snap frozen on dry ice once harvested. The tissue was then kept in -80 oC until further processing.
2.3 RNA Isolation and Quality Assessment
The muscles were homogenised with an IKA® T10 basic Homogenizer (IKA, USA) and RNA was isolated using the MasterPure™ RNA Purification Kit (Epicentre, USA) with the additional RNase-Free DNase I (Epicentre, USA) step according to the manufacturer's protocol. Quality and quantification of RNA was measured with the NanoVue™ Plus Spectrophotometer (GE Healthcare, UK) and the integrity confirmed by visualisation on agarose gel electrophoresis. Additional quality and quantification assessment was performed prior microarray analysis with the Agilent 2100 Bioanalyzer system on the Bioanalyzer RNA 6000 Nano chip according to the Eukaryote Total RNA Nano assay protocol (Agilent, USA).
2.4 Microarray sample preparation and analysis
Sample preparation and hybridisation kits from Agilent were used to process each RNA samples and RNA samples were hybridised onto Agilent-SurePrint G3 Mouse GE 8x60K Microarray chip (Agilent, U.S.A) following the manufacturer's protocol. The samples were prepared with the RNA Spike-In Kit, One-Color Kit (Agilent, USA) according to the manufacturer's protocol. The samples were then labelled and amplified with the Low Input Quick Amp Labeling Kit, One-Color (Agilent, USA) generating fluorescent complementary RNA (cRNA). The cRNA samples were then purified with the RNeasy Mini Kit (Qiagen, Germany). The purified cRNA was quantified using the NanoVue™ Plus Spectrophotometer (GE Healthcare, UK). The Cyanine 3 dye concentration (pmol/μl), RNA absorbance ration (260 nm/280 nm) and cRNA concentration (ng/μl) were recorded. The yield and specific activity of each reaction was determined and only samples with minimum yield and specific activity of 0.825 μg and 6 pmol Cy3 per μg of cRNA, respectively, were selected for hybridization onto the chip using the Gene Expression Hybridization Kit and Hybridization Gasket Slide Kit (Agilent, USA) according to the manufacturer's protocol. The array with the hybridization mix with samples was incubated in the Agilent SureHyb chamber hybridized at 60oC for 17 hours while rotating. After hybridization, the slides were washed with the Gene Expression Wash Buffer Kit (Agilent, USA), with the addition of Triton X-102 to the wash buffers. The slides were scanned with the Agilent SureScan Microarray Scanner. The Agilent Feature Extraction (FE) program was used to extract the information from probe features from the scanned microarray data. Information from the images (.tif) are extracted where the FE program assigns a default grid template and protocol automatically for each extraction set. The QC report generated is checked to confirm that the microarray has been successful.
2.5 Analysis of Differential Gene Expression
The microarray expression data extracted with the FE program was analysed with the Agilent GeneSpring Software v.13.1. The data was imported into the software and normalised by percentile shift normalisation at 75th percentile. This is a global normalisation method which adjusts the location of all the spot intensities in the array. Each column is taken independently and the nth percentile of the expression values for this array is computed across all spots, ranging from 0 to 100 and n = 50 as the median. The value is the subtracted from the expression value of each entity. Following this, the entities are filtered by expression. The cut-off percentile used was lower cut-off = 20th percentile and upper cut-off of 100th percentile. The microarray expression data sets were compared between two groups namely wild-type and Ts1Cje with each muscle group data analysed separately. The data was statistically analysed using moderated t-Test with Westfall Young Permutative multiple testing correction. Genes were considered significantly differentially expressed when the corrected p-values ≤ 0.05 and absolute fold change value of ≥1.5.
2.6 Functional clustering of DEGs
The DEGs were clustered based on functional ontologies using the Database for Annotation, Visualisation and Integrated Discovery (DAVID) software [37]. Stringent classification criteria (a kappa similarity threshold of 0.50, a minimum term overlap of three, three initial and final group memberships with a 0.50 multiple linkage threshold and a modified Fisher-exact p-value on enrichment thresholds of 1.0) was employed in the analysis. The DEGs in each cluster and common genes among the clusters were identified. The list of DEGs for soleus and EDL were compared to determine the genes found on MMU16 as well as those not on MMU16 which were differentially regulated in both muscles.
2.7 RT-qPCR
Complementary DNA (cDNA) was synthesised from 500 ng of high quality total RNA using the Transcriptor First Strand cDNA Synthesis Kit (Roche, Switzerland) with anchored-oligo(dT)18 primers according to manufacturer's protocol. The optional step of denaturing the template-primer mixture was performed. Quantitative PCR was performed using the LightCycler® 480 Real-Time PCR System (Roche, Switzerland). RT-qPCR was performed using LightCycler® 480 SYBR Green I Master (Roche, Switzerland) at the following conditions: pre-incubation at 95oC (5 minutes), followed by 45 cycles of 95oC (10 seconds), 60oC (10 seconds) and 72oC (10 seconds) and melting analysis from 65oC to 97oC with continuous signal acquisitions (every +0.2oC). The reaction was then cooled down to 40oC. Relative quantification by standard curve method was performed according to the protocol published previously [38] with the normalisation step performed against any 2 of the 3 of the housekeeping genes; phosphoglycerate kinase 1 (Pgk1), hydroxymethylbilane synthase (Hmbs) and proteasome subunit beta type-2 (Psmb2). See Table 1 for list of primers used in the analysis.
Table 1: List of primers used for RT-qPCR assays
|
Gene |
Forward Primer (5'→3') |
Reverse Primer (5'→3') |
Amplicon Size (nt) |
|
Itsn1 |
accagttctcgcctgatgat |
aggcggaccacaatctctc |
18 |
|
Rcan1 |
cccgacaaacagttcctcat |
catgcagttcatacttctctcc |
52 |
|
Synj1 |
ctcctgacagccaaagcaa |
ggcttcagtggttctggaag |
18 |
|
Cdk13 |
gcaactcaaatgtagcacctgt |
ttgccaattcactgtggttta |
66 |
|
Donson |
tgttggcctgtctctggataa |
agcactggcctctgactga |
111 |
|
Dyrk1a |
tcagtcttcaggcaccacct |
tgttactcgttcccgaggat |
65 |
|
Ifnar1 |
gtgcagtgtataagcaccacagt |
ggacatagctcttgccttgg |
85 |
|
Ifnar2 |
tgagcaggatgcgttcac |
tctctaggctcgcagacacc |
81 |
|
Mansc1 |
ttggcaactcaggaagactg |
cgggtgtcaaagatcatcaa |
92 |
|
Runx1 |
acatcgggaattccttcaca |
tcgcttcacctcattcacag |
91 |
|
Sod1 |
caggacctcattttaatcctcac |
tgcccaggtctccaacat |
78 |
|
Tmem50b |
ccggactgaggttgatcg |
catggagttctcattaaacaatgg |
126 |
|
Pgk1 |
tacctgctggctggatgg |
cacagcctcggcatatttct |
65 |
|
Psmb2 |
gagggcagtggagcttctta |
aggtgggcagattcaagatg |
71 |
|
Hmbs |
aaagttccccaacctggaat |
ccaggacaatggcactgaat |
98 |
2.8 Western blotting
The muscle samples were homogenized in ice-cold RIPA buffer (Millipore Corporation, USA) supplemented with a protease inhibitor cocktail (Calbiochem, USA) and phosphatase inhibitors (Calbiochem, USA). A total of 20 μg of protein lysate was separated on 10% SDS-PAGE and transferred to PVDF membranes (BioRad). After blocking with 5% non-fat dried milk, the membrane was incubated with mouse monoclonal anti-Gapdh (Abcam, UK; 1:1000 dilution; Catalog ID: ab125247), mouse monoclonal anti-Ifnar1 (Biolegend, USA; 1:500 dilution; Catalog ID: 127322) or goat polyclonal anti-Ifnar2 (Santa Cruz, USA; 1:500 dilution; Catalog ID: sc-20218) primary antibodies at 4oC overnight, followed by goat anti-mouse IgG (Santa Cruz, USA; 1:1000 dilution; Catalog ID: sc-2005) or donkey anti-goat (Santa Cruz, USA; 1:1000 dilution for anti-Ifnar and 1:2500 for anti-Gapdh; Catalog ID: sc-2020) HRP-conjugated secondary antibodies at room temperature for 1 hour. Immunoreactivity of the proteins were detected and visualised using the Advansta WesternBrightTM SiriusTM chemiluminescence kit (Advansta Corp, USA). Images were taken with a G:BOX F3 Gel Documentation Workstation (Syngene, UK) and pixelation analysis of the bands were performed using ImageJ (http://imagej.nih.gov/ij/) according to the standard protocol. The density of the bands were quantified and normalised against the loading control (housekeeping protein), glyceraldehyde 3-phosphate dehydrogenase (Gapdh).
3. RESULTS
3.1 Differentially Expressed Genes (DEGs)
To investigate the effect of partial trisomy on muscle weakness in Ts1Cje, we performed a microarray analysis on soleus and EDL (n=4 per tissue per group) muscles between Ts1Cje and wildtype disomic littermates using the SurePrint G3 Mouse GE 8x60K Microarray chip (Agilent Technologies, USA). Following filtering and normalisation, a total of 40,932 probes were detected for soleus with 10,316 of them being long non-coding (lnc) RNA whereas 42,399 probes were detected in EDL with 10,959 of them being lncRNA. The expression datasets are publicly accessible from the Gene Expression Omnibus database repository under the series accession number GSE96572. Figure 1a depicts the summary of differentially expressed probes and genes in the skeletal muscles. The differentially expressed probes were selected based on corrected p-values ≤ 0.05 and absolute fold change value of ≥1.5 (Figure 1b). Considering only the coding genes, 166 DEGs and 262 DEGs were identified in the soleus and EDL muscles, respectively. This suggests that the partial trisomy of MMU16 in Ts1Cje mice has a greater effect on gene expression in EDL. Of these DEGs, 35 in soleus and 9 in EDL were trisomic genes found in the Ts1Cje mice (Table 2; Figure 1c; Table S1). When the list of DEGs of both skeletal muscle types were compared, only 6 trisomic DEGs (A930006K02Rik, Donson, Dyrk1a, Runx1, Sod1 and Tmem50b) and 3 disomic DEGs (Cdk13, Dnah11 and Mansc1) were common or consistently dysregulated in both the soleus and EDL (Figure 1c).
The dynein axonemal heavy chain 11 (Dnah11) gene stood out among the DEGs found with a significant upregulation in both soleus and EDL muscles of the Ts1Cje mice with an approximately 460- and 550-fold change in EDL and soleus, respectively (Table S1). The upregulation of Dnah11 is consistent with previously reported microarray expression results of the brain [39,40] and is probably specific to the Ts1Cje mouse mode [41]. This is due to the partial monosomy of MMU12 resulting from a partial translocation of MMU16 onto Dnah11 locus leading to a segmental deletion of the gene in the Ts1Cje mouse model [39,42]. Thus, Dnah11 was inferred to be uninvolved in molecular contributions to hypotonia in DS as it is specific to Ts1Cje and not translated to a functional protein resulting from the translocation. While the observation does not have significant to muscle weakness in DS, presence of this overexpression served as a validation of the results from the microarray analysis.
3.2 Functional analysis of DEGs
To determine the disrupted molecular networks at a global level, a top-down functional clustering approach using DAVID [37] was performed. The analysis revealed 5 functional clusters in soleus associated with signal transduction (13 DEGs, 5 ontologies), development of reproductive system (4 DEGs, 6 ontologies), nucleic acid biosynthesis (6 DEGs, 12 ontologies), protein modification and metabolism (13 DEGs, 4 ontologies) as well as regulation of gene expression (20 DEGs, 21 ontologies) (Table 3). From the 45 DEGs clustered, 9 DEGs namely Atp5o, Drd4, Fcer1a, Gm8566, Lyn, Morc3, Pawr, Sod1 and Tlr3 were found to be clustered into more than one functional annotation cluster. On the other hand, only 3 functional clusters were revealed for EDL namely neuron and cell development (11 DEGs, 14 ontologies), protein modification and metabolic processes (26 DEGs, 5 ontologies) as well as ion transport (10 DEGs, 14 ontologies) as listed in Table 3. From the 42 DEGs clustered, 7 of those DEGs namely Atp6v1e2, Cdh4, Gm8566, Ipmk, Runx1, Sod1 and Tgfbr1 were found to be clustered into more than one functional annotation cluster.
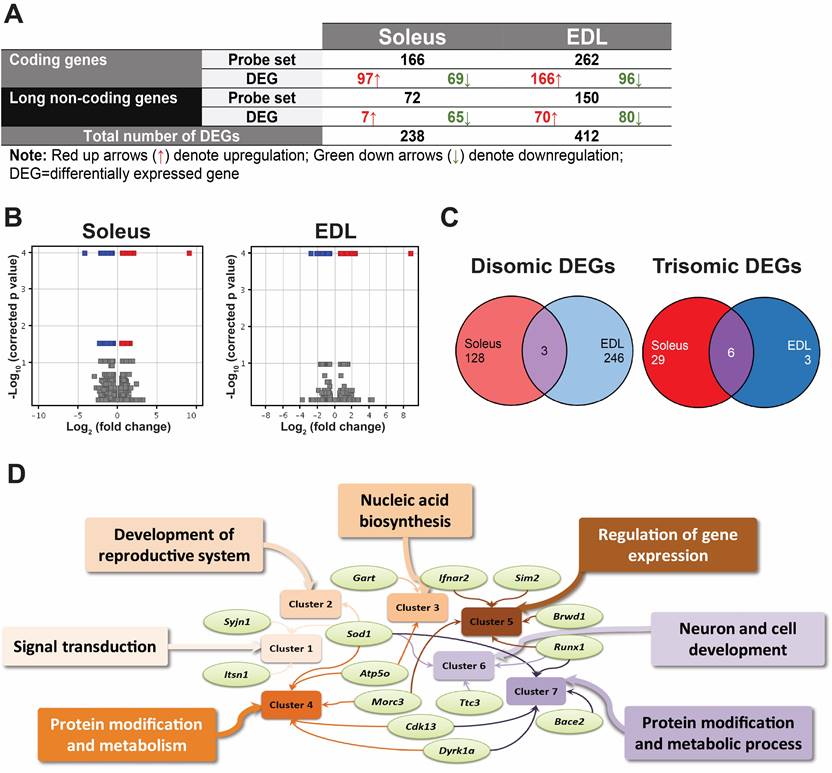
Figure 1. Figure 1 Analysis of differentially expressed genes (DEGs). (a) Summary of differentially expressed genes (DEGs) in both muscles. (b) Volcano plots for differentially expressed probes for soleus and EDL. (c) Venn diagrams depict the distribution of disomic and trisomic DEGs in both muscles. (d) Significant functional clusters associated with trisomic DEGs based on DAVID analysis.
Table 2: List of trisomic differentially expressed genes (DEGs)
|
Soleus |
|||
|
Probe Name |
Full Gene Name |
Gene Symbol |
Log2 Expression of Ts1Cje-WT |
|
A_52_P447284 |
Chloride intracellular channel 6 |
Clic6 |
2.0793 |
|
A_52_P236705 |
Ripply3 homolog (zebrafish) |
Ripply3 |
1.4131 |
|
A_51_P514085 |
Myxovirus (influenza virus) resistance 2 |
Mx2 |
1.2231 |
|
A_55_P1981739 |
Downstream neighbor of SON |
Donson |
1.0974 |
|
A_55_P2177539 |
Superoxide dismutase 1, soluble |
Sod1 |
1.0932 |
|
A_55_P2101920 |
Eva-1 homolog C (C. Elegans) |
Eva1c |
1.0822 |
|
A_55_P2183438 |
Runt related transcription factor 1 |
Runx1 |
1.0619 |
|
A_55_P2206461 |
RIKEN cDNA A930006K02 gene |
A930006K02Rik |
0.9124 |
|
A_51_P313483 |
Dual-specificity tyrosine-(Y)-phosphorylation regulated kinase 1a |
Dyrk1a |
0.8927 |
|
A_51_P510849 |
Transmembrane protein 50B |
Tmem50b |
0.8537 |
|
A_52_P190405 |
Interferon (alpha and beta) receptor 2 |
Ifnar2 |
0.8372 |
|
A_52_P301223 |
Bromodomain and WD repeat domain containing 1 |
Brwd1 |
0.7939 |
|
A_55_P2042600 |
Uncharacterized LOC102641028 |
LOC102641028 |
0.7870 |
|
A_55_P1955517 |
Synaptojanin 1 |
Synj1 |
0.7825 |
|
A_51_P314521 |
Small integral membrane protein 11 |
Smim11 |
0.7658 |
|
A_51_P481238 |
Dopey family member 2 |
Dopey2 |
0.7588 |
|
A_51_P508191 |
Down syndrome critical region gene 3 |
Dscr3 |
0.7534 |
|
A_55_P2190426 |
Single-minded homolog 2 (Drosophila) |
Sim2 |
0.7457 |
|
A_51_P160907 |
Microrchidia 3 |
Morc3 |
0.7450 |
|
A_55_P2181109 |
Interleukin 10 receptor, beta |
Il10rb |
0.7287 |
|
A_66_P100232 |
PAX3 and PAX7 binding protein 1 |
Paxbp1 |
0.7148 |
|
A_52_P58257 |
Mitochondrial ribosomal protein S6 |
Mrps6 |
0.6874 |
|
A_55_P1952788 |
Son DNA binding protein |
Son |
0.6823 |
|
A_55_P1952793 |
Son DNA binding protein |
Son |
0.6681 |
|
A_55_P2032297 |
RIKEN cDNAB230307C23 gene |
B230307C23Rik |
0.6676 |
|
A_55_P2171158 |
High mobility group nucleosomal binding domain 1 |
Hmgn1 |
0.6601 |
|
A_66_P134728 |
Proteasome (prosome, macropain) assembly chaperone 1 |
Psmg1 |
0.6596 |
|
A_55_P2091671 |
Proteasome (prosome, macropain) assembly chaperone 1 |
Psmg1 |
0.6582 |
|
A_55_P2033135 |
SH3-binding domain glutamic acid-rich protein |
Sh3bgr |
0.6579 |
|
A_55_P2055682 |
ATP synthase, H+ transporting, mitochondrial F1 complex, O subunit |
Atp5o |
0.6551 |
|
A_52_P639774 |
Phosphoribosylglycinamide formyltransferase |
Gart |
0.6464 |
|
A_55_P1952915 |
SH3-binding domain glutamic acid-rich protein |
Sh3bgr |
0.6441 |
|
A_55_P1994586 |
Intersectin 1 (SH3 domain protein 1A) |
Itsn1 |
0.6323 |
|
A_55_P1977985 |
phosphatidylinositol glycan anchor biosynthesis, class P |
Pigp |
0.6274 |
|
A_51_P196395 |
RIKEN cDNA 1110004E09 gene |
1110004E09Rik |
0.6076 |
|
EDL |
|||
|
Probe Name |
Full Gene Name |
Gene Symbol |
Log2 Expression of Ts1Cje-WT |
|
A_55_P2438722 |
Interferon (alpha and beta) receptor 1 |
Ifnar1 |
1.2305 |
|
A_51_P481221 |
Beta-site APP-cleaving enzyme 2 |
Bace2 |
1.1339 |
|
A_55_P2177539 |
Superoxide dismutase 1, soluble |
Sod1 |
0.8570 |
|
A_55_P2206461 |
RIKEN cDNA A930006K02 gene |
A930006K02Rik |
0.7994 |
|
A_51_P510849 |
Transmembrane protein 50B |
Tmem50b |
0.7763 |
|
A_55_P1981739 |
Downstream neighbor of SON |
Donson |
0.7702 |
|
A_55_P2143195 |
Dual-specificity tyrosine-(Y)-phosphorylation regulated kinase 1a |
Dyrk1a |
0.7598 |
|
A_55_P2183438 |
Runt related transcription factor 1 |
Runx1 |
0.7216 |
|
A_66_P106065 |
Tetratricopeptide repeat domain 3 |
Ttc3 |
0.6111 |
These results for soleus and EDL highlighted dysregulation of genes involved in basic molecular function suggesting that muscle weakness in Ts1Cje is not caused by genes involved directly in muscle development and function but by indirect gene dosage effect on signal transmission and basic cell functions in the muscles. Trisomic DEGs implicated in all these clusters regardless of muscle types were Atp5o, Bace2, Brwd1, Cdk13, Dyrk1a, Gart, Ifnar2, Itsn1, Morc3, Runx1, Sim2, Sod1, Synj1 and Ttc3 (Figure 1d). Overall, the analysis indicated that protein modification and metabolism (Clusters 4 and 7) and regulation of gene expression (Cluster 5) functional clusters affected by the greatest number of trisomic DEGs.
Table 3: Functional clustering and gene ontology of differentially expressed genes (DEGs) in soleus and extensor digitorum longus (EDL) muscles
|
Soleus |
||
|
Functional Cluster |
Genes |
Gene ontologies |
|
Cluster 1 Signal Transduction (Enrichment Score: 1.2217) |
Drd4, Fcer1a, Fgd3, Gm8566, Itsn1, Lyn, Pawr, Rgn, Sod1, Sos1, Synj1, Tlr3, Wwox |
GO:0009966~Regulation Of Signal Transduction GO:0009967~Positive Regulation Of Signal Transduction GO:0010647~Positive Regulation Of Cell Communication GO:0010627~Regulation Of Protein Kinase Cascade GO:0010740~Positive Regulation Of Protein Kinase Cascade GO:0007242~Intracellular Signalling Cascade |
|
Cluster 2 Development Of Reproductive System (Enrichment Score: 1.1991) |
Gm8566, Lepr, Msh4, Sod1 |
GO:0022602~Ovulation Cycle Process GO:0008585~Female Gonad Development GO:0046545~Development Of Primary Female Sexual Characteristics GO:0046660~Female Sex Differentiation GO:0008406~Gonad Development GO:0045137~Development Of Primary Sexual Characteristics |
|
Cluster 3 Nucleic Acid Biosynthesis (Enrichment Score: 1.1433) |
Atp13a4, Atp1b4, Atp5o, Cox17, Fignl1, Gart |
GO:0009150~Purine Ribonucleotide Metabolic Process GO:0009144~Purine Nucleoside Triphosphate Metabolic Process GO:0009260~Ribonucleotide Biosynthetic Process GO:0006164~Purine Nucleotide Biosynthetic Process GO:0042625~Atpase Activity, Coupled To Transmembrane Movement Of Ions GO:0006753~Nucleoside Phosphate Metabolic Process GO:0034654~Nucleobase, Nucleoside, Nucleotide And Nucleic Acid Biosynthetic Process GO:0034404~Nucleobase, Nucleoside And Nucleotide Biosynthetic Process GO:0042626~Atpase Activity, Coupled To Transmembrane Movement Of Substances GO:0009142~Nucleoside Triphosphate Biosynthetic Process GO:0015405~P-P-Bond-Hydrolysis-Driven Transmembrane Transporter Activity GO:0006812~Cation Transport |
|
Cluster 4 Protein Modification & Metabolism (Enrichment Score: 0.7400) |
Atp5o, Bub1b, Camk4, Cdk13, Dyrk1a, Fcer1a, Gm8566, Lyn, Morc3, Pigp, Sod1, Stk35, Uevldv
|
GO:0016310~Phosphorylation GO:0004672~Protein Kinase Activity GO:0006464~Protein Modification Process GO:0044267~Cellular Protein Metabolic Process |
|
Cluster 5 Regulation Of Gene Expression (Enrichment Score: 0.5947) |
Brwd1, Dna2, Drd4, Elk4, Fcer1a, Foxe3, Gng7, Hdx, Hoxd8, Hoxd9, Icosl, Ifnar2, Lyn, Mcm8, Morc3,Nr5a2, Pawr, Runx1, Sim2, Tlr3
|
GO:0042108~Positive Regulation Of Cytokine Biosynthetic Process GO:0010604~Positive Regulation Of Macromolecule Metabolic Process GO:0031325~Positive Regulation Of Cellular Metabolic Process GO:0010557~Positive Regulation Of Macromolecule Biosynthetic Process GO:0031328~Positive Regulation Of Cellular Biosynthetic Process GO:0042035~Regulation Of Cytokine Biosynthetic Process GO:0009891~Positive Regulation Of Biosynthetic Process GO:0002694~Regulation Of Leukocyte Activation GO:0031326~Regulation Of Cellular Biosynthetic Process GO:0019219~Regulation Of Nucleobase, Nucleoside, Nucleotide And Nucleic Acid Metabolic Process GO:0010556~Regulation Of Macromolecule Biosynthetic Process GO:0006355~Regulation Of Transcription, DNA-Dependent GO:0051252~Regulation Of RNA Metabolic Process GO:0045935~Positive Regulation Of Nucleobase, Nucleoside, Nucleotide And Nucleic Acid Metabolic Process GO:0051173~Positive Regulation Of Nitrogen Compound Metabolic Process GO:0045449~Regulation Of Transcription GO:0010468~Regulation Of Gene Expression GO:0045893~Positive Regulation Of Transcription, DNA-Dependent GO:0051254~Positive Regulation Of RNA Metabolic Process GO:0045941~Positive Regulation Of Transcription GO:0010628~Positive Regulation Of Gene Expression |
|
EDL |
||
|
Functional Cluster |
Genes |
Gene ontologies |
|
Cluster 6 Neuron & Cell Development (Enrichment Score: 0.7211) |
Cdh4, Dpysl5, Efna5, Gm8566, Hoxa1, Ipmk, Runx1, Sod1, Sostdc1, Tgfbr1, Ttc3 |
GO:0048667~Cell Morphogenesis Involved In Neuron Differentiation GO:0048666~Neuron Development GO:0000904~Cell Morphogenesis Involved In Differentiation GO:0048699~Generation Of Neurons GO:0022008~Neurogenesis GO:0007409~Axonogenesis GO:0048812~Neuron Projection Morphogenesis GO:0048858~Cell Projection Morphogenesis GO:0000902~Cell Morphogenesis GO:0032990~Cell Part Morphogenesis GO:0031175~Neuron Projection Development GO:0048568~Embryonic Organ Development GO:0048562~Embryonic Organ Morphogenesis GO:0009887~Organ Morphogenesis |
|
Cluster 7 Protein Modification & Metabolic Process (Enrichment Score: 0.5895) |
Acsm3, Asb1, Atp6v1e2, Bace2, Cdk13, Csk, Dhx58, Dyrk1a, Erbb4, Gm8566, Hspa13, Ipmk, Isg15, Loc100046931, Map3k10, Mmaa, Msh5, Runx1, Sod1, St6galnac6, St8sia6, Stk35, Tcp10b, Tcp10c, Tdrd9, Tgfbr1 |
GO:0016310~Phosphorylation GO:0032559~Adenyl Ribonucleotide Binding GO:0004672~Protein Kinase Activity GO:0006464~Protein Modification Process GO:0044267~Cellular Protein Metabolic Process |
|
Cluster 8 Ion Transport (Enrichment Score: 0.5084) |
Atp6v1e2, Cdh4,Kcne3, Kcnmb1, Kctd19, P2ry12, Slc12a5, Slc30a7, Slc V34a1, Tnfrsf13b |
GO:0015672~Monovalent Inorganic Cation Transport GO:0034705~Potassium Channel Complex GO:0008076~Voltage-Gated Potassium Channel Complex GO:0034703~Cation Channel Complex GO:0006812~Cation Transport GO:0030001~Metal Ion Transport GO:0034702~Ion Channel Complex GO:0008324~Cation Transmembrane Transporter Activity GO:0005887~Integral To Plasma Membrane GO:0031226~Intrinsic To Plasma Membrane GO:0031420~Alkali Metal Ion Binding GO:0022836~Gated Channel Activity GO:0005216~Ion Channel Activity GO:0022838~Substrate Specific Channel Activity |
3.3 Quantitative PCR validation of selected DEGs
RT-qPCR was used to validate the DEGs identified in the microarray analysis. The genes selected for validation were found to be differentially regulated in both skeletal muscles (Cdk13 and Mansc1), trisomic genes or reported in previous studies (Itsn1, Rcan1, Synj1, Donson, Dyrk1a, Ifnar1, Ifnar2, Runx1, Sod1 and Tmem50b). Only trisomic genes found to be upregulated in both soleus and EDL were selected for validation. This is to focus on genes found to be dysregulated regardless of muscle type. In total, all genes except for Cdk13, were validated by RT-qPCR using the same RNA that was used for the microarray analyses, which consisted of soleus and EDL skeletal muscle tissues from Ts1Cje (n=6) and disomic mice (n=7) (Figure 2). The expression profile of a gene was considered validated when both microarray and RT-qPCR data showed a consistent directional change. Our results showed that all trisomic genes were upregulated in the Ts1Cje samples with fold differences ≥ 1.50.
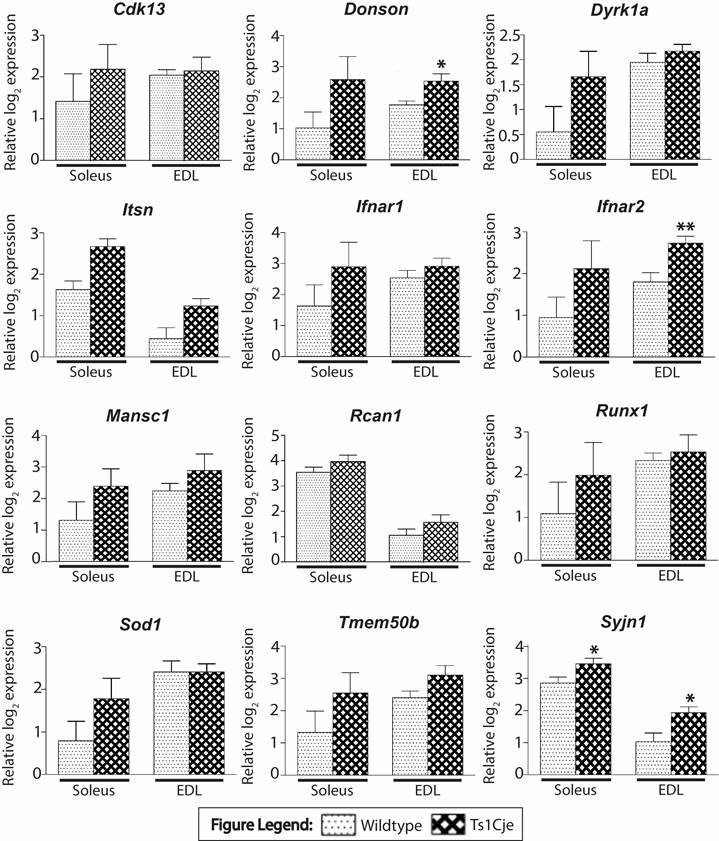
Figure 2. RT-qPCR validation of DEGs. Comparison of expression profiles of selected DEG candidates in soleus and EDL muscles between Ts1Cje and wildtype groups. Graph bars represents relative log2 mean ∓ standard error of mean (normalized against any 2 of the 3 housekeeping genes: Pgk1, Psmb2 or Hmbs). Asterisks denote the statistical significance at p-value˂0.05 (*) or ˂0.01 (**) based on Student's t-test.
Among the trisomic genes dysregulated, Ifnar1 and Ifnar2 were found to be upregulated in the soleus and EDL of Ts1Cje, respectively. Thus, Western blot analysis was performed to evaluate the effect of mRNA levels on protein synthesis of the interferon receptors (Figure 3a). Based on the pixelation analysis of the non-saturated bands (Figure 3), Ifnar1 and Ifnar2 protein expressions were found to be lower in the Ts1Cje soleus by 2.10- and 1.72-fold, respectively (Figure 3b). In the EDL of the Ts1Cje, Ifnar2 protein expression was found to be lower in the Ts1Cje mice when compared to wild type by 1.43-fold. The finding was in contrary to the results obtained from microarray and RT-qPCR analysis where the genes were upregulated in Ts1Cje mice.
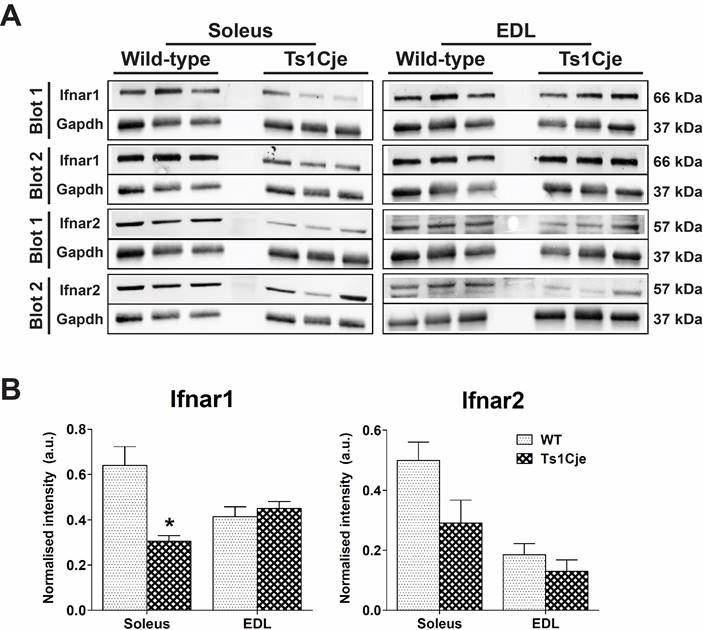
Figure 3. Western blot analysis of Ifnar1 and Ifnar2. (a) Ifnar1 (66 kDa) and Ifnar2 (57 kDa) protein expression in both soleus and EDL muscles (n=3 per tissue per group) of Ts1Cje and wild-type mice. Gapdh protein expression (37 kDa) was used to normalise any loading errors. Blot 1 and Blot 2 represent independent technical replicates. (b) Densitometry analysis of the bands in (a) depicting the normalised level of Ifnar1 and Ifnar 2 in the soleus and EDL muscles. The asterisk denotes the statistical significance at p-value ˂0.05 (*) based on Student's t-test.
4. DISCUSSION
Hypotonia is one of the clinical phenotypes found in DS individuals and the actual underlying cause of hypotonia in DS remains unknown. Defects within the central or peripheral nervous systems, transmission of signals at the neuromuscular junctions or the muscles itself can be attributed to hypotonia. The underlying cause of these defects however, lies within the effect of additional genetic material in DS. Multiple theories have been formed on the link between the trisomy of HSA21 to the phenotypes such as the dosage imbalance hypothesis suggesting a 50% increase in expression of genes on chromosome 21q [2,43]; the amplified developmental instability hypothesis proposes that the trisomy causes general alteration in development homeostasis leading to the phenotypes [43-45]; and the DSCR hypothesis predicting that triplication of the region was sufficient to cause DS phenotypes [46]. Thus, this study aims to elucidate the potential cause of hypotonia in DS individuals through global transcriptomic gene expression study to identify genes potentially involved in Ts1Cje muscle weakness.
There are limited studies involving the characterisation of the transcriptomic profile in Ts1Cje skeletal muscle especially according to the muscle type. To our knowledge, this is the first attempt to profile on the transcriptomes of Ts1Cje soleus and EDL muscles. Our findings showed that the gene expression profile between the soleus and EDL muscles were different with more DEGs found in the EDL muscles. A higher number of DEGs were found in EDL suggesting that the fast-twitch muscle may be more affected in Ts1Cje as compared to the slow-twitch muscle, soleus. The current study focused on the DEGs found to be significantly dysregulated in both muscles. This was to further investigate the potential role of the DEGs in muscle weakness while ensuring that the difference in expression was not due to muscle type.
The microarray results showed a significant increase expression of 35 trisomic genes in soleus and 6 trisomic genes in EDL by at least 1.5-fold when compared to their disomic littermates. The data set had similar results to previous studies on soleus muscle in Ts65Dn [47] as well as studies on brains and primary neuro stem cells and progenitor cells of Ts1Cje mice [40,48-51] where there was a significant dosage-dependent over-expression of the genes located on the triplicated region of MMU16. Collectively, the DEGs in the Ts1Cje soleus and EDL were mainly involved in basic molecular function such as signal transduction, protein modification and metabolism, nucleic acid biosynthesis, and regulation of gene expression in agreement with previous studies on Ts65Dn mice [52] and human samples [53,54] suggesting that muscle weakness in Ts1Cje was not caused by genes involved directly in muscle development and function but by indirect gene dosage effect on signal transmission and basic cell functions in the muscles.
Studies have suggested that muscle weakness is a result of defects in the CNS of DS patients. Several neuroanatomical defects affecting the cerebellum and cerebral cortex have been reported in DS individuals [22,55-57]. However, motor function involves both the nervous and muscular systems. Defective neuromuscular junction is one of the contributing elements in causing muscle weakness where two trisomic genes Itsn1 and Synj1 were found to be overexpressed in Ts1Cje. These genes are critical regulators of endocytosis, synaptic transmission, and cell signalling [58,59]. Overexpression of Itsn1 has been linked to neurodegeneration in mice [60] and Drosophila [61] while dysregulation of Synj1 leads to severe weakness, ataxia, spontaneous epileptic seizures, and poor motor coordination [62,63] as well as abnormal synaptic morphology at the neuromuscular junctions in Drosophila homologs of HSA21 [59]. Synj1 has also been linked to Parkinson's disease where mutations of this gene were found to be associated with the disease [64,65]. Thus upregulation of these genes may promote neurodegeneration and interference of signal transmission which in turn affects motor function as supported by previous studies [60-63].
Runx1 and Sod1 were clustered in more than one functional annotation cluster indicating its role in muscle weakness. However, in the Ts1Cje mice, Sod1 is part of the proximal break point of the translocated segment of MMU16 to MMU12 causing one of the triplicated copies of Sod1 gene to be non-functional [29,48]. While this is unique to Ts1Cje, Sod1 has consistently been identified as a marker for DS in different models where it is upregulated. Although Ts1Cje has 100% rather than 150% CuZnSOD activity [29], the consistent upregulation of the mRNA in Ts1Cje model suggests the need to investigate the level of Sod1 expression or CuZnSOD activity in other DS models presented with muscle weakness phenotype.
A high number of genes involved in the inflammatory response such as cytokines, interleukins and interferon receptors were found to be differentially regulated in Ts1Cje skeletal muscles. Studies have shown that the inflammatory response plays a role in tissue healing and thus is important for skeletal muscle regeneration after injury [66-69]. Runx1, another trisomic gene, was also found to be upregulated in Ts1Cje skeletal muscles. Interestingly, Runx1 also known as AML1 was first identified in acute myeloid leukemia and has been shown to regulate hematopoiesis, angiogenesis, muscle function, and neurogenesis [70-73]. Runx1 was shown to be crucial in muscle regeneration and a decrease in this gene lead to severe muscle atrophy [74] as implicated in Duchenne muscular dystrophy (DMD), myopathy patients [75] and myasthenia gravis [76] suggesting a role in the immune response. A study showed that overexpression of Runx1 triggered inflammation [77] suggesting that upregulation of Runx1 in DS may be linked to hypotonia by decreasing efficiency of muscle regeneration as a respond to excessive anti-inflammatory process.
Rcan1 or Dscr1 was found to be upregulated in Ts1Cje muscles. Expression of RCAN1 was found in the brain, skeletal and cardiac muscle, and the expression was elevated in brains of Alzheimer's disease patients and Down syndrome foetuses [78-80]. RCAN1-calcineurin-SOD1 pathway was activated during high oxidative stress leading to less cardiovascular risk in normal individuals [81] whereas increased RCAN1 has been found to reduce cancer risk in Down syndrome individuals [82] or reduced tumour growth incidence in Ts65Dn mainly by suppressing the growth of blood vessels that innervate the area [83]. The role of Rcan1 and its upregulation in Ts1Cje muscles remains unknown and it would be intriguing to suggest that upregulated Rcan1 may have reduced the blood supply to muscles leading to perturbed growth or regeneration associated with weaknesses seen in Ts1Cje model.
Interestingly, expression of two interferon receptors namely Ifnar1 and Ifnar2 at the mRNA and protein levels in Ts1Cje muscles had contrasting results. The trisomic genes were upregulated in Ts1Cje as expected, but the proteins were found to be downregulated in the muscles. The observation suggests an incomplete penetrance of gene dosage effect at least in the skeletal muscle as compared to the adult brain of Ts1Cje [40]. While the observation could be attributed to post-transcriptional or translational regulation and differences in the tissue being analysed, IFN activity has been shown to regulate proliferation of muscles cells especially by triggering the JAK-STAT signalling pathway [84]. Downregulation of Ifnar1 and Ifnar2 are expected to reduce JAK-STAT signalling pathway which in turn would promote myoblast renewal and regeneration processes [85]. However, a previous study of skeletal muscle stem cells (SMSCs) derived from Ts1Cje and control mice of the same age did not show any differences in the number SMSC as well as their differentiation capability in vitro [86]. Although the finding suggests no apparent differences in myogenesis during normal physiological state of Ts1CJe muscles, it does not preclude the potential involvement of Ifnar1 and Ifnar2 via JAK-STAT signalling pathway during early skeletal muscle development or regeneration state which will become obvious in aging subjects or during muscle injuries.
Other trisomic genes such as Dyrk1a, Donson and Tmem50b were also found upregulated in the brain of Ts1Cje mouse model [40]. Dyrk1a is a well-known DS marker, and mouse models with overexpressed Dyrk1a transgenes exhibited motor abnormalities [26] that were comparable to DS patients [87]. The role of Dyrk1a during muscle development and function has not been well documented but its expression has been shown to be regulated by myocyte specific enhancer 2d (Mef2d), a transcription factor that is important for muscle cell differentiation [88]. On the other hand, no relevant reports were available on the role of Donson and Tmem50b in the skeletal muscle function and their contributions to muscle weakness in Ts1Cje should be investigated further. Similarly, the disomic DEG Mansc1, was found to be upregulated in both soleus and EDL but no prior study has linked the role of the gene to muscle development or function. Further characterisation of these under-documented novel candidate genes may provide invaluable information on the progression of muscle weakness in Ts1Cje or hypotonia in DS individuals.
5. CONCLUSIONS
To the best of our knowledge, there has not been a study to determine the gene profiles of both Type I and Type II skeletal muscles in Ts1Cje mice. This transcriptomic profiling of Ts1Cje skeletal muscles led to the identification of trisomic DEGs that are implicated in signal transmission and basic cell functions, through impairment of neuromusclular junction signalling as well as oxidative stress and chronic inflammation. This provides new insights on the potential disrupted molecular pathways associated with muscle weakness in Ts1Cje and can help elucidate the cause for hypotonia in DS individuals. However, only a subset of genes and proteins were selected for validation. Therefore, additional genes including the non-coding genes should be validated in the future. To further study the role of the dysregulated genes in hypotonia, gain- or loss-of-function studies should be performed to determine the role of selected DEGs on the development, function and regeneration of skeletal muscles. Downstream or upstream targets of the genes in the molecular pathways should also be studied to provide more insight to the underlying mechanism involved.
Supplementary Materials: Table S1: List of all DEGs and Table S2: List of trisomic DEGs are available online at https://www.neuroscirn.org/ojs/index.php/nrnotes/article/view/12.
Acknowledgements: This work was supported in part by funding from the MOHE Fundamental Research Grant Scheme (04-01-15-1663FR), UPM Geran Putra IPT (UPM/700/2/1/GP-IPT/2013/9409500) and UPM Geran Putra IPS (UPM/700/2/1/GP-IPS/2014/9448800) awarded to PSC; UPM Research University Grant Scheme (04-02-12-2102RU) and MOSTI ScienceFund (02-01-04-SF2336) awarded to KHL. MPYL and CLL were recipients of the Malaysian Ministry of Higher Education MyBrain scholarship.
Author Contributions: MPYL, UB and CLL conceived the study, carried out the experiments and analysed the data, and drafted the manuscript. MPYL, KHL, PSC and RR conceived the study, participated in its design and coordination, and wrote the manuscript. All authors read and approved the final manuscript.
Conflicts of Interest: The authors declare no conflict of interest.
References
1. Epstein CJ, Korenberg JR, Annerén G, Antonarakis SE, Aymé S, Courchesne E, et al. Protocols to establish genotype-phenotype correlations in Down syndrome. Am J Hum Genet. 1991;49(1):207-235. https://www.ncbi.nlm.nih.gov/pubmed/1829580
2. Antonarakis SE, Lyle R, Dermitzakis ET, Reymond A, Deutsch S. Chromosome 21 and down syndrome: from genomics to pathophysiology. Nat Rev Genet. 2004;5(10):725-738. https://doi.org/10.1038/nrg1448
3. Rigoldi C, Galli M, Mainardi L, Crivellini M, Albertini G. Postural control in children, teenagers and adults with Down syndrome. Res Dev Disabil. 2011;32(1):170-175. https://doi.org/10.1016/j.ridd.2010.09.007
4. Jover M, Ayoun C, Berton C, Carlier M. Specific grasp characteristics of children with trisomy 21. Dev Psychobiol. 2010;52(8):782-793. https://doi.org/10.1002/dev.20474
5. Malak R, Kotwicka M, Krawczyk-Wasielewska A, Mojs E, Samborski W. Motor skills, cognitive development and balance functions of children with Down syndrome. Ann Agric Environ Med. 2013;20(4):803-806. https://www.ncbi.nlm.nih.gov/pubmed/24364457
6. Vicari S. Motor development and neuropsychological patterns in persons with Down syndrome. Behav Genet. 2006;36(3):355-364. https://doi.org/10.1007/s10519-006-9057-8
7. Virji-Babul N, Kerns K, Zhou E, Kapur A, Shiffrar M. Perceptual-motor deficits in children with Down syndrome: implications for intervention. Downs Syndr Res Prac. 2006;10(2):74-82. htps://doi.org/10.3104/reports.308
8. Mon-Williams M, Tresilian JR, Bell VE, Coppard VL, Jobling A, Carson RG. The preparation of reach to grasp movements in adults with Down syndrome. Hum Mov Sci. 2001;20(4-5):587-602. https://doi.org/10.1016/S0167-9457(01)00069-0
9. Brown R, Taylor J, Matthews B. Quality of life--ageing and Down syndrome. Downs Syndr Res Pract. 2001;6(3):111-116. https://doi.org/10.3104/case-studies.101
10. Kraemer BR, McIntyre LL, Blacher J. Quality of life for young adults with mental retardation during transition. Taylor SJ, editor. Ment Retard. 2003;41(4):250-262. https://doi.org/10.1352/0047-6765(2003)41<250:QOLFYA>2.0.CO;2
11. Cunningham C. Families of children with Down syndrome. Downs Syndr Res Pract. 1996;4(3):87-95. https://doi.org/10.3104/perspectives.66
12. Cioni M, Cocilovo A, Di Pasquale F, Araujo MB, Siqueira CR, Bianco M. Strength deficit of knee extensor muscles of individuals with Down syndrome from childhood to adolescence. Am J Ment Retard. 1994;99(2):166-174. https://www.ncbi.nlm.nih.gov/pubmed/7803033
13. Horvat M, Croce R, Pitetti KH, Fernhall B. Comparison of isokinetic peak force and work parameters in youth with and without mental retardation. Med Sci Sports Exerc. 1999;31(8):1190-1195. https://www.ncbi.nlm.nih.gov/pubmed/10449023
14. Mercer VS, Lewis CL. Hip Abductor and Knee Extensor Muscle Strength of Children with and without Down Syndrome. Pediatr Phys Ther. 2001;13(1):18-26. https://www.ncbi.nlm.nih.gov/pubmed/17053646
15. Henderson SE, Morris J, Frith U. The motor deficit in Down's syndrome children: A problem of timing? J Child Psychol Psychiatry. 1981;22(3):233-245. https://doi.org/10.1111/j.1469-7610.1981.tb00549.x
16. Anson JG. Chapter 13 Neuromotor Control and Down Syndrome. In: Approaches to the Study of Motor Control and Learning. Elsevier; 1992. pp. 387-412. (Advances in Psychology; vol. 84). https://doi.org/10.1016/S0166-4115(08)61693-3
17. Latash ML, Almeida GL, Corcos DM. Preprogrammed reactions in individuals with Down syndrome: the effects of instruction and predictability of the perturbation. Arch Phys Med Rehabil. 1993;74(4):391-399. https://www.ncbi.nlm.nih.gov/pubmed/8466421
18. Riquelme Agulló I, Manzanal González B. Motor development in children with Down syndrome and associated osteoarticular pathology. Int Med Rev Down Syndr. 2006;10(3):34-40. https://doi.org/10.1016/S2171-9748(06)70067-8
19. Kesslak JP, Nagata SF, Lott I, Nalcioglu O. Magnetic resonance imaging analysis of age-related changes in the brains of individuals with Down's syndrome. Neurology. 1994;44(6):1039-1039. https://doi.org/10.1212/WNL.44.6.1039
20. Haydar TF, Reeves RH. Trisomy 21 and early brain development. Trends Neurosci. 2012;35(2):81-91. https://doi.org/10.1016/j.tins.2011.11.001
21. Riquelme Agulló I, Manzanal González B. Factors influencing motor development in children with Down syndrome. Int Med Rev Down Syndr. 2006;10(2):18-24. https://doi.org/10.1016/S2171-9748(06)70063-0
22. Pinter JD, Eliez S, Schmitt JE, Capone GT, Reiss AL. Neuroanatomy of Down's syndrome: a high-resolution MRI study. Am J Psychiatry. 2001;158(10):1659-1665. https://doi.org/10.1176/appi.ajp.158.10.1659
23. Aylward EH, Habbak R, Warren AC, Pulsifer MB, Barta PE, Jerram M, et al. Cerebellar volume in adults with Down syndrome. Arch Neurol. 1997;54(2):209-212. https://doi.org/10.1001/archneur.1997.00550140077016
24. Davisson MT, Schmidt C, Akeson EC. Segmental trisomy of murine chromosome 16: a new model system for studying Down syndrome. Prog Clin Biol Res. 1990;360:263-280. https://www.ncbi.nlm.nih.gov/pubmed/2147289
25. Galante M, Jani H, Vanes L, Daniel H, Fisher EMC, Tybulewicz VLJ, et al. Impairments in motor coordination without major changes in cerebellar plasticity in the Tc1 mouse model of Down syndrome. Hum Mol Genet. 2009;18(8):1449-1463. https://doi.org/10.1093/hmg/ddp055
26. Altafaj X, Dierssen M, Baamonde C, Martí E, Visa J, Guimerà J, et al. Neurodevelopmental delay, motor abnormalities and cognitive deficits in transgenic mice overexpressing Dyrk1A (minibrain), a murine model of Down's syndrome. Hum Mol Genet. 2001;10(18):1915-1923. https://www.ncbi.nlm.nih.gov/pubmed/11555628
27. Costa AC, Walsh K, Davisson MT. Motor dysfunction in a mouse model for Down syndrome. Physiol Behav. 1999;68(1-2):211-220. https://doi.org/10.1016/S0031-9384(99)00178-X
28. Gardiner K. Gene-dosage effects in Down syndrome and trisomic mouse models. Genome Biol. 2004;5(10):244. https://doi.org/10.1186/gb-2004-5-10-244
29. Sago H, Carlson EJ, Smith DJ, Kilbridge J, Rubin EM, Mobley WC, et al. Ts1Cje, a partial trisomy 16 mouse model for Down syndrome, exhibits learning and behavioral abnormalities. Proc Natl Acad Sci USA. 1998;95(11):6256-6261. https://doi.org/10.1073/pnas.95.11.6256
30. Reeves RH, Irving NG, Moran TH, Wohn A, Kitt C, Sisodia SS, et al. A mouse model for Down syndrome exhibits learning and behaviour deficits. Nat Genet. 1995;11(2):177-184. https://doi.org/10.1038/ng1095-177
31. Sago H, Carlson EJ, Smith DJ, Rubin EM, Crnic LS, Huang TT, et al. Genetic dissection of region associated with behavioral abnormalities in mouse models for Down syndrome. Pediatr Res. 2000;48(5):606-613. https://doi.org/10.1203/00006450-200011000-00009
32. Siarey RJ, Villar AJ, Epstein CJ, Galdzicki Z. Abnormal synaptic plasticity in the Ts1Cje segmental trisomy 16 mouse model of Down syndrome. Neuropharmacology. 2005;49(1):122-128. https://doi.org/10.1016/j.neuropharm.2005.02.012
33. Pennington BF, Moon J, Edgin J, Stedron J, Nadel L. The neuropsychology of Down syndrome: evidence for hippocampal dysfunction. Child Dev. 2003;74(1):75-93. https://doi.org/10.1111/1467-8624.00522
34. Belichenko PV, Kleschevnikov AM, Salehi A, Epstein CJ, Mobley WC. Synaptic and cognitive abnormalities in mouse models of Down syndrome: exploring genotype-phenotype relationships. J Comp Neurol. 2007;504(4):329-345. https://doi.org/10.1002/cne.21433
35. Bareja A, Holt JA, Luo G, Chang C, Lin J, Hinken AC, et al. Human and mouse skeletal muscle stem cells: convergent and divergent mechanisms of myogenesis. Alway SE, editor. PLoS ONE. 2014;9(2):e90398. https://doi.org/10.1371/journal.pone.0090398
36. Scott W, Stevens J, Binder-Macleod SA. Human skeletal muscle fiber type classifications. Phys Ther. 2001;81(11):1810-1816. https://doi.org/10.1093/ptj/81.11.1810
37. Huang DW, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009;37(1):1-13. https://doi.org/10.1093/nar/gkn923
38. Ling KH, Hewitt CA, Beissbarth T, Hyde L, Banerjee K, Cheah P-S, et al. Molecular networks involved in mouse cerebral corticogenesis and spatio-temporal regulation of Sox4 and Sox11 novel antisense transcripts revealed by transcriptome profiling. Genome Biol. 2009;10(10):R104. https://doi.org/10.1186/gb-2009-10-10-r104
39. Laffaire J, Rivals I, Dauphinot L, Pasteau F, Wehrle R, Larrat B, et al. Gene expression signature of cerebellar hypoplasia in a mouse model of Down syndrome during postnatal development. BMC Genomics. 2009;10(1):138. https://doi.org/10.1186/1471-2164-10-138
40. Ling KH, Hewitt CA, Tan K-L, Cheah P-S, Vidyadaran S, Vidyadaran S, et al. Functional transcriptome analysis of the postnatal brain of the Ts1Cje mouse model for Down syndrome reveals global disruption of interferon-related molecular networks. BMC Genomics. 2014;15(1):624. https://doi.org/10.1186/1471-2164-15-624
41. Amano K, Sago H, Uchikawa C, Suzuki T, Kotliarova SE, Nukina N, et al. Dosage-dependent over-expression of genes in the trisomic region of Ts1Cje mouse model for Down syndrome. Hum Mol Genet. 2004;13(13):1333-1340. https://doi.org/10.1093/hmg/ddh154
42. Duchon A, Raveau M, Chevalier C, Nalesso V, Sharp AJ, Herault Y. Identification of the translocation breakpoints in the Ts65Dn and Ts1Cje mouse lines: relevance for modeling Down syndrome. Mamm Genome. 2011;22(11-12):674-684. https://doi.org/10.1007/s00335-011-9356-0
43. Pritchard MA, Kola I. The "gene dosage effect" hypothesis versus the "amplified developmental instability" hypothesis in Down syndrome. J Neural Transm Suppl. 1999;57:293-303. https://www.ncbi.nlm.nih.gov/pubmed/10666684
44. Shapiro BL. Down syndrome--a disruption of homeostasis. Am J Med Genet. 1983;14(2):241-269. https://doi.org/10.1002/ajmg.1320140206
45. Moldrich RX. A yeast model of Down syndrome. Int J Dev Neurosci. 2007;25(8):539-543. https://doi.org/10.1016/j.ijdevneu.2007.10.001
46. Belichenko NP, Belichenko PV, Kleschevnikov AM, Salehi A, Reeves RH, Mobley WC. The "Down Syndrome Critical Region" Is Sufficient in the Mouse Model to Confer Behavioral, Neurophysiological, and Synaptic Phenotypes Characteristic of Down Syndrome. J Neurosci. 2009;29(18):5938-5948. https://doi.org/10.1523/JNEUROSCI.1547-09.2009
47. Cowley PM, Keslacy S, Middleton FA, DeRuisseau LR, Fernhall B, Kanaley JA, et al. Functional and biochemical characterization of soleus muscle in Down syndrome mice: insight into the muscle dysfunction seen in the human condition. Am J Physiol Regul Integr Comp Physiol. 2012;303(12):R1251-1260. https://doi.org/10.1152/ajpregu.00312.2012
48. Amano K, Sago H, Uchikawa C, Suzuki T, Kotliarova SE, Nukina N, et al. Dosage-dependent over-expression of genes in the trisomic region of Ts1Cje mouse model for Down syndrome. Hum Mol Genet. 2004;13(13):1333-1340. https://doi.org/10.1093/hmg/ddh154
49. Potier M-C, Rivals I, Mercier G, Ettwiller L, Moldrich RX, Laffaire J, et al. Transcriptional disruptions in Down syndrome: a case study in the Ts1Cje mouse cerebellum during post-natal development. J Neurochem. 2006;97 Suppl 1(s1):104-109. https://doi.org/10.1111/j.1471-4159.2005.03624.x
50. Hewitt CA, Ling KH, Merson TD, Simpson KM, Ritchie ME, King SL, et al. Gene network disruptions and neurogenesis defects in the adult Ts1Cje mouse model of Down syndrome. Aziz SA, editor. PLoS ONE. 2010;5(7):e11561. https://doi.org/10.1371/journal.pone.0011561
51. Tan K-L, Ling KH, Hewitt CA, Cheah P-S, Simpson K, Gordon L, et al. Transcriptional profiling of the postnatal brain of the Ts1Cje mouse model of Down syndrome. Genomics Data. 2014;2:314-317. https://doi.org/10.1016/j.gdata.2014.09.009
52. Cowley PM. Skeletal Muscle Function, Morphology, and Biochemistry in Ts65Dn Mice: A Model of Down Syndrome. Exercise Science - Doctoral Dissertations. Syracuse University. December 2011. https://surface.syr.edu/ppe_etd/5.
53. Lockstone HE, Harris LW, Swatton JE, Wayland MT, Holland AJ, Bahn S. Gene expression profiling in the adult Down syndrome brain. Genomics. 2007;90(6):647-660. https://doi.org/10.1016/j.ygeno.2007.08.005
54. Vilardell M, Rasche A, Thormann A, Maschke-Dutz E, Pérez-Jurado LA, Lehrach H, et al. Meta-analysis of heterogeneous Down Syndrome data reveals consistent genome-wide dosage effects related to neurological processes. BMC Genomics. 2011;12(1):229. https://doi.org/10.1186/1471-2164-12-229
55. Anderson JS, Nielsen JA, Ferguson MA, Burback MC, Cox ET, Dai L, et al. Abnormal brain synchrony in Down Syndrome. Neuroimage Clin. 2013;2:703-715. https://doi.org/10.1016/j.nicl.2013.05.006
56. Guidi S, Bonasoni P, Ceccarelli C, Santini D, Gualtieri F, Ciani E, et al. Neurogenesis impairment and increased cell death reduce total neuron number in the hippocampal region of fetuses with Down syndrome. Brain Pathol. 2008;18(2):180-197. https://doi.org/10.1111/j.1750-3639.2007.00113.x
57. Guidi S, Ciani E, Bonasoni P, Santini D, Bartesaghi R. Widespread proliferation impairment and hypocellularity in the cerebellum of fetuses with down syndrome. Brain Pathol. 2011;21(4):361-373. https://doi.org/10.1111/j.1750-3639.2010.00459.x
58. Wong KA, Wilson J, Russo A, Wang L, Okur MN, Wang X, et al. Intersectin (ITSN) family of scaffolds function as molecular hubs in protein interaction networks. Aguilar RC, editor. PLoS ONE. 2012;7(4):e36023. https://doi.org/10.1371/journal.pone.0036023
59. Chang KT, Min K-T. Upregulation of three Drosophila homologs of human chromosome 21 genes alters synaptic function: implications for Down syndrome. Proc Natl Acad Sci USA. 2009;106(40):17117-17122. https://doi.org/10.1073/pnas.0904397106
60. Gelderblom M, Eminel S, Herdegen T, Waetzig V. c-Jun N-terminal kinases (JNKs) and the cytoskeleton--functions beyond neurodegeneration. Int J Dev Neurosci. 2004;22(7):559-564. https://doi.org/10.1016/j.ijdevneu.2004.07.014
61. Scappini E, Koh T-W, Martin NP, O'Bryan JP. Intersectin enhances huntingtin aggregation and neurodegeneration through activation of c-Jun-NH2-terminal kinase. Hum Mol Genet. 2007;16(15):1862-1871. https://doi.org/10.1093/hmg/ddm134
62. Cremona O, Di Paolo G, Wenk MR, Lüthi A, Kim WT, Takei K, et al. Essential role of phosphoinositide metabolism in synaptic vesicle recycling. Cell. 1999;99(2):179-188. https://doi.org/10.1016/S0092-8674(00)81649-9
63. Harris TW, Hartwieg E, Horvitz HR, Jorgensen EM. Mutations in synaptojanin disrupt synaptic vesicle recycling. J Cell Biol. 2000;150(3):589-600. https://doi.org/10.1083/jcb.150.3.589
64. Quadri M, Fang M, Picillo M, Olgiati S, Breedveld GJ, Graafland J, et al. Mutation in the SYNJ1 gene associated with autosomal recessive, early-onset Parkinsonism. Hum Mutat. 2013;34(9):1208-1215. https://doi.org/10.1002/humu.22373
65. Drouet V, Lesage S. Synaptojanin 1 mutation in Parkinson's disease brings further insight into the neuropathological mechanisms. BioMed Res Int. 2014;2014(48):289728-289729. https://doi.org/10.1155/2014/289728
66. Arnold L, Henry A, Poron F, Baba-Amer Y, van Rooijen N, Plonquet A, et al. Inflammatory monocytes recruited after skeletal muscle injury switch into antiinflammatory macrophages to support myogenesis. J Exp Med. 2007;204(5):1057-1069. https://doi.org/10.1084/jem.20070075
67. Bondesen BA, Mills ST, Kegley KM, Pavlath GK. The COX-2 pathway is essential during early stages of skeletal muscle regeneration. Am J Physiol Cell Physiol. 2004;287(2):C475-483. https://doi.org/10.1152/ajpcell.00088.2004
68. Bryer SC, Fantuzzi G, van Rooijen N, Koh TJ. Urokinase-type plasminogen activator plays essential roles in macrophage chemotaxis and skeletal muscle regeneration. J Immunol. 2008;180(2):1179-1188. https://doi.org/10.4049/jimmunol.180.2.1179
69. Lescaudron L, Peltékian E, Fontaine-Pérus J, Paulin D, Zampieri M, Garcia L, et al. Blood borne macrophages are essential for the triggering of muscle regeneration following muscle transplant. Neuromuscul Disord. 1999;9(2):72-80. https://doi.org/10.1016/S0960-8966(98)00111-4
70. Biggs JR, Peterson LF, Zhang Y, Kraft AS, Zhang D-E. AML1/RUNX1 phosphorylation by cyclin-dependent kinases regulates the degradation of AML1/RUNX1 by the anaphase-promoting complex. Mol Cell Biol. 2006;26(20):7420-7429. https://doi.org/10.1128/MCB.00597-06
71. Xavier AC, Taub JW. Acute leukemia in children with Down syndrome. Haematologica. 2010;95(7):1043-1045. https://doi.org/10.3324/haematol.2010.024968
72. Zhu X, Yeadon JE, Burden SJ. AML1 is expressed in skeletal muscle and is regulated by innervation. Mol Cell Biol. 1994;14(12):8051-8057. https://doi.org/10.1128/MCB.14.12.8051
73. Edwards H, Xie C, LaFiura KM, Dombkowski AA, Buck SA, Boerner JL, et al. RUNX1 regulates phosphoinositide 3-kinase/AKT pathway: role in chemotherapy sensitivity in acute megakaryocytic leukemia. Blood. 2009;114(13):2744-2752. https://doi.org/10.1182/blood-2008-09-179812
74. Wang X, Blagden C, Fan J, Nowak SJ, Taniuchi I, Littman DR, et al. Runx1 prevents wasting, myofibrillar disorganization, and autophagy of skeletal muscle. Genes Dev. 2005;19(14):1715-1722. https://doi.org/10.1101/gad.1318305
75. Umansky KB, Gruenbaum-Cohen Y, Tsoory M, Feldmesser E, Goldenberg D, Brenner O, et al. Runx1 Transcription Factor Is Required for Myoblasts Proliferation during Muscle Regeneration. Cox GA, editor. PLoS Genet. 2015;11(8):e1005457. https://doi.org/10.1371/journal.pgen.1005457
76. Zhou Y, Kaminski HJ, Gong B, Cheng G, Feuerman JM, Kusner L. RNA expression analysis of passive transfer myasthenia supports extraocular muscle as a unique immunological environment. Invest Ophthalmol Vis Sci. 2014;55(7):4348-4359. https://doi.org/10.1167/iovs.14-14422
77. Luo M-C, Zhou S-Y, Feng D-Y, Xiao J, Li W-Y, Xu C-D, et al. Runt-related Transcription Factor 1 (RUNX1) Binds to p50 in Macrophages and Enhances TLR4-triggered Inflammation and Septic Shock. J Biol Chem. 2016;291(42):22011-22020. https://doi.org/10.1074/jbc.M116.715953
78. Porta S, Serra SA, Huch M, Valverde MA, Llorens F, Estivill X, et al. RCAN1 (DSCR1) increases neuronal susceptibility to oxidative stress: a potential pathogenic process in neurodegeneration. Hum Mol Genet. 2007;16(9):1039-1050. https://doi.org/10.1093/hmg/ddm049
79. Mitchell AN, Jayakumar L, Koleilat I, Qian J, Sheehan C, Bhoiwala D, et al. Brain expression of the calcineurin inhibitor RCAN1 (Adapt78). Arch Biochem Biophys. 2007;467(2):185-192. https://doi.org/10.1016/j.abb.2007.08.030
80. Sobrado M, Ramirez BG, Neria F, Lizasoain I, Arbones ML, Minami T, et al. Regulator of calcineurin 1 (Rcan1) has a protective role in brain ischemia/reperfusion injury. J Neuroinflammation. 2012;9(1):48. https://doi.org/10.1186/1742-2094-9-48
81. Chávez CJ, Ortega P, D'Escrivan A, Miranda LE, Leal M JY, Delgado C. Body mass index changes and lipid peroxidation in adults with Down's syndrome. Int Med Rev Down Syndr. 2012;16(2):19-25. https://doi.org/10.1016/S2171-9748(12)70021-1
82. Liu F, Liang Z, Wegiel J, Hwang Y-W, Iqbal K, Grundke-Iqbal I, et al. Overexpression of Dyrk1A contributes to neurofibrillary degeneration in Down syndrome. FASEB J. 2008;22(9):3224-3233. https://doi.org/10.1096/fj.07-104539
83. Baek K-H, Zaslavsky A, Lynch RC, Britt C, Okada Y, Siarey RJ, et al. Down's syndrome suppression of tumour growth and the role of the calcineurin inhibitor DSCR1. Nature. 2009;459(7250):1126-1130. https://doi.org/10.1038/nature08062
84. Price FD, Maltzahn von J, Bentzinger CF, Dumont NA, Yin H, Chang NC, et al. Inhibition of JAK-STAT signaling stimulates adult satellite cell function. Nat Med. 2014;20(10):1174-1181. https://doi.org/10.1038/nm.3655
85. Doles JD, Olwin BB. The impact of JAK-STAT signaling on muscle regeneration. Nat Med. 2014;20(10):1094-1095. https://doi.org/10.1038/nm.3720
86. Lim CL, Bala U, Leong MP-Y, Stanslas J, Ramasamy R, Ling K-H, et al. Cellular function of satellite cells does not play a role in muscle weakness of adult Ts1Cje mice. Neurosci Res Notes. 2018;1(1):3-10. https://doi.org/10.31117/neuroscirn.v1i1.6
87. Vicari S, Bellucci S, Carlesimo GA. Visual and spatial long-term memory: differential pattern of impairments in Williams and Down syndromes. Dev Med Child Neurol. 2005;47(5):305-311. https://doi.org/10.1111/j.1469-8749.2005.tb01141.x
88. Wang P, Wang L, Chen L, Sun X. Dual-specificity tyrosine-phosphorylation regulated kinase 1A Gene Transcription is regulated by Myocyte Enhancer Factor 2D. Sci Rep. 2017;7(1):7240. https://doi.org/10.1038/s41598-017-07655-1